Green Energy & Water Electrolysis
The transition to renewable energy requires efficient storage technologies. Water electrolysis — splitting H₂O into hydrogen and oxygen using electricity — is a cornerstone of this transition. Understanding and optimizing the underlying catalytic reactions is a central scientific challenge.
The Energy & Climate Challenge
Despite scientific consensus on anthropogenic climate change, global energy production in 2019 still relied overwhelmingly on fossil fuels. Atmospheric CO₂ concentrations are at historic highs — a trend confirmed by Antarctic ice-core records stretching back 800,000 years.
Renewable sources (solar, wind, hydro) are growing rapidly, but their intermittent nature creates a fundamental challenge: energy must be stored when supply exceeds demand and released when it does not.
- Electrochemical storage offers high energy density and scalability
- Hydrogen gas is a clean energy vector when produced electrolytically
- Round-trip efficiency depends critically on catalyst overpotential
- Li-ion batteries, pumped hydro, and H₂ each cover different power/capacity regimes
Energy Storage Technologies
Relative capacity/applicability of current storage technologies
Relative suitability of energy storage technologies by scale and deployment flexibility
Water Electrolysis
First demonstrated in 1789 by van Troostwijk and Deiman, water electrolysis decomposes liquid water into gaseous hydrogen and oxygen using electrical energy. The thermodynamic requirement is 2.46 eV per water molecule (237.2 kJ/mol), corresponding to 1.23 V per electron transfer.
The overall reaction proceeds through two half-cell reactions at the electrodes:
Cathode (HER)
4H⁺ + 4e⁻ → 2H₂
Low overpotential on most metals
Anode (OER)
2H₂O → O₂ + 4H⁺ + 4e⁻
Rate-limiting step, high overpotential
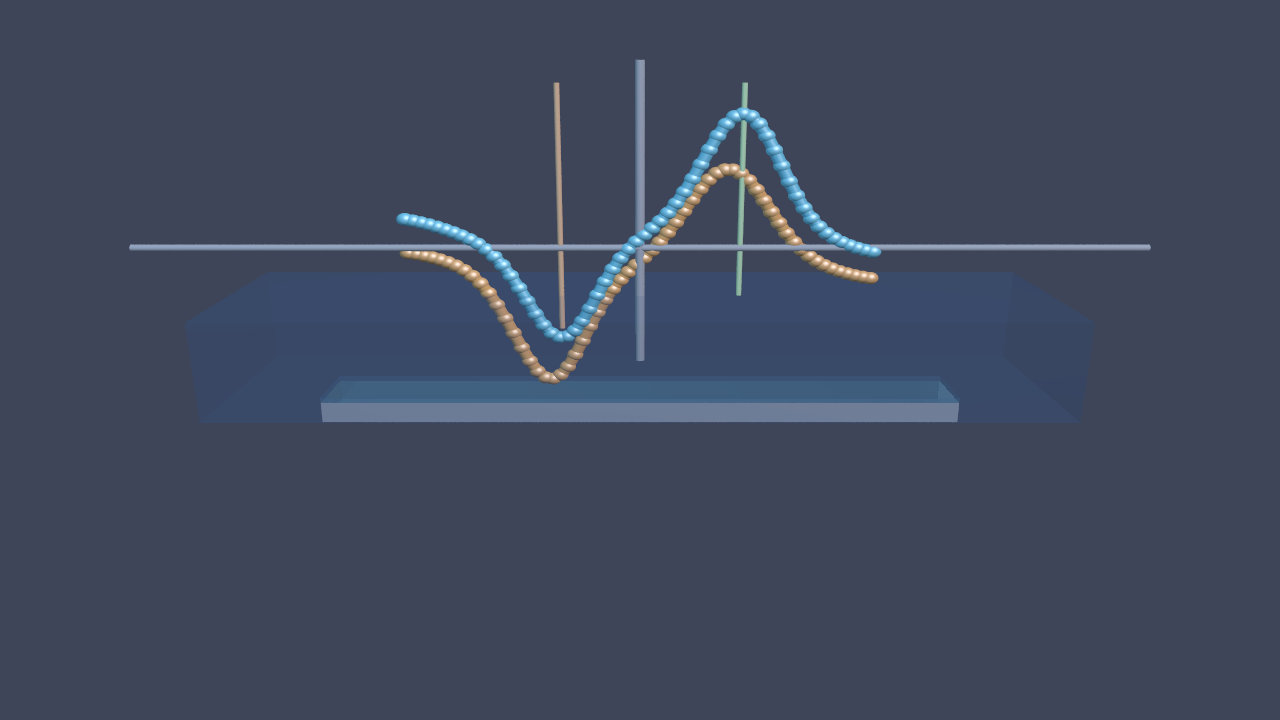
Schematic CV showing anodic (blue) and cathodic (orange) sweeps with characteristic peaks
OER Kinetics & the Volcano Plot
The oxygen evolution reaction (OER) involves four sequential proton-coupled electron transfers, each ideally requiring 1.23 V. Any additional voltage needed — the overpotential η — represents an energy loss.
The four-step OER mechanism on a metal oxide surface:
H₂O + * → *OH + H⁺ + e⁻Water dissociation and OH adsorption
*OH → *O + H⁺ + e⁻Dehydrogenation to adsorbed oxygen
*O + H₂O → *OOH + H⁺ + e⁻Often the rate-limiting step on most catalysts
*OOH → O₂ + H⁺ + e⁻ + *Desorption regenerates surface site
The hydrogen evolution reaction (HER) is simpler — a two-step Volmer–Heyrovsky or Volmer–Tafel mechanism — and proceeds with much lower overpotential on most metallic catalysts.
The Brønsted–Evans–Polanyi (BEP) relation states that kinetic barriers for surface reactions scale linearly with adsorption energies. This allows us to compute theoretical overpotentials from DFT adsorption energies alone — orders of magnitude faster than computing full transition states.
Assuming BEP scaling holds, the descriptor ΔG*O − ΔG*OH captures the competition between oxygen binding strength (too strong → *OOH formation limited) and too-weak binding (→ *OH dehydrogenation limited).
Plotting theoretical overpotential against the OER descriptor (ΔG*O − ΔG*OH) for a range of transition metal oxides produces the characteristic volcano plot. Materials at the peak — IrO₂ and RuO₂ — bind oxygen intermediates optimally.
- Left flank: Strong O binding — *OOH formation is rate-limiting
- Right flank: Weak O binding — *OH → *O dehydrogenation is rate-limiting
- Volcano peak: IrO₂ and RuO₂ achieve lowest theoretical overpotential (~0.4 V)
- TiO₂: Located far right — too weak O binding, η ≈ 1.2 V
Catalyst Materials
IrO₂
Industrial gold standard for acidic OER. Binds oxygen intermediates optimally — close to the volcano peak. Stable in acidic conditions but based on scarce Ir.
RuO₂
Slightly more active than IrO₂ but dissolves ~10× faster at 1.2 V in acidic media. Investigates as comparison material in surface phase diagram studies.
Ti–IrO₂ Alloy
Mixing Ti into the IrO₂ host can modify adsorption energies and improve OER activity. Computationally designed — DFT identifies optimal Ti motifs and surface configurations.
High-Entropy Oxides
Recent work explores multi-component oxides (5+ elements) that access vast composition spaces. ML-driven screening and in-situ XAS characterize active sites.